

Su nombre es Melissa Vásquez Cerdas, tiene 42 años y es de las pocas especialistas costarricenses encargadas de diagnosticar por vías moleculares uno de los padecimientos hereditarios más raros de la medicina registrados en el mundo: la enfermedad de Huntington.
Esta académica e investigadora realiza su labor en el Instituto de Investigaciones en Salud de la Universidad de Costa Rica (Inisa-UCR), en la Sección de Genética. Ahí, actualmente y con la ayuda de la técnica de laboratorio Dayana Vargas Sanabria, se efectúan los análisis que le permite a un paciente saber, finalmente, si tiene o no esa extraña enfermedad que degenera de forma paulatina algunas de las células nerviosas del cerebro.
“La enfermedad de Huntington es un padecimiento genético hereditario y neurodegenerativo, causado por un daño o mutación en una parte importante del material genético (ADN). La enfermedad es muy poco frecuente y tiene una prevalencia entre cinco a diez personas afectadas por cada 100 000 a nivel mundial. En Costa Rica, desde el 2004 en el Inisa-UCR se ha realizado el diagnóstico molecular a 162 personas. De esa cifra, solo 70 tenían la mutación causante de la enfermedad de Huntington y, de esos, según tenemos conocimiento 20 ya fallecieron”, comentó Vásquez.
Gracias al trabajo de esta científica, su carisma por su profesión en el campo de la biología molecular y al compromiso continuo del Instituto, Costa Rica es de los pocos países de Centroamérica capaces de brindarle a su población una opción para diagnosticar genéticamente esta enfermedad sin ningún fin de lucro.
“Esta patología tiene una connotación psicosocial muy importante vinculada a la gravedad y a la irreversibilidad de los síntomas, así como a una manifestación usualmente tardía que causa gran impacto a quienes la padecen. Este tipo de enfermedad constituye un desafío para el paciente, su familia y el sistema de salud, porque por su poca frecuencia implica mayores esfuerzos para diagnosticarla de forma temprana, comprender sus manifestaciones clínicas y encontrar los tratamientos indicados. Si bien el número de personas con esta condición es reducido, desde la visión humanista de la UCR cada una merece que su afección sea contemplada”, ahondó Vásquez.
El padecimiento de Huntington fue descrito por primera vez durante el año 1872 por el médico estadounidense George Huntington. Él proporcionó la descripción clásica de la enfermedad después de estudiar a varias generaciones de una familia que experimentó síntomas parecidos.
La historia de este padecimiento engloba grandes curiosidades y misterios. Por esa razón, la M.Sc. Melissa Vásquez Cerdas brindó un rato de su tiempo para aclarar con lujo de detalle la presencia de esta enfermedad en el país, el proceso diagnóstico, los tratamientos y hasta cómo pasó de considerarse una enfermedad ocasionada por espíritus malignos a un padecimiento clínico debidamente documentado por la ciencia.

Los diagnósticos moleculares o genéticos son muy importantes pero no pueden predecir la progresión de la enfermedad. Tampoco, cuáles van a ser los síntomas específicos de la persona ni cuándo empezarán. En la fotografía está Dayana Vargas realizando el procesamiento de la muestra.
Contexto
-La enfermedad de Huntington es una de las enfermedades más infrecuentes en el mundo, ¿por qué se interesó en estudiarla, Melissa?
Melissa Vásquez Cerdas (MVC): “Desde finales de los noventas, en el Inisa-UCR había una línea de investigación de enfermedades neurológicas causadas por mutaciones conocidas como ‘inestables’. El término 'inestable' se refiere a que el tamaño de la mutación que las causa varía.
Quien inició esa línea de estudio fue la Dra. Patricia Cuenca, investigadora y exdirectora del Inisa-UCR. Ella empezó con el síndrome del cromosoma X frágil. Posteriormente, el Dr. Fernando Morales comenzó a estudiar la distrofia miotónica tipo I, cuyos estudios continúan hasta la fecha. Por supuesto que son enfermedades distintas, pero lo que comparten es que son causadas por un tipo de mutación similar (mutaciones inestables).
La enfermedad de Huntington empieza a ser contemplada cuando entré al Inisa como asistente y se me dio la posibilidad de hacer mi tesis de maestría en la misma línea de investigación de enfermedades neurológicas causadas por mutaciones inestables. Entonces, se me propuso incorporar la enfermedad de Huntington. Cerca del 2002 empezamos a realizar los primeros experimentos con el propósito de implementar y poner en práctica el diagnóstico molecular para este padecimiento.
En el 2004 se planteó un proyecto concreto de investigación y, a partir de ese año, ya teníamos la implementación del diagnóstico que fue lo que, justamente, consistió mi proyecto de tesis de maestría en biología con énfasis en genética y biología molecular”.
-Entonces, ¿antes del año 2000 ya se sabía que en Costa Rica existía esa enfermedad?
MVC:“Antes del 2004, en la revisión de literatura científica que se hizo en ese momento, encontramos una publicación de 1974 relacionada con la enfermedad. Fue publicada por el Dr. Arnaldo Antillón del Hospital San Juan de Dios, en donde él hace un reporte de caso clínico de una niña con antecedentes familiares de la enfermedad."
-Ahora bien, Melissa, ¿por qué este tipo de mutación es considerada como “inestable”? ¿Qué nos dice esto en comparación con el resto de las mutaciones?
MVC: “Las mutaciones inestables consisten en repeticiones de tripletas. Todas estas enfermedades (síndrome del cromosoma X frágil, la distrofia miotónica tipo I y la enfermedad de Huntington) son causadas por una expansión de tripletas de los componentes del ADN. Recordemos que el ADN posee cuatro bases: citosina (C), adenina (A), timina (T) y guanina (G).
En el caso de la distrofia miotónica tipo 1, la mutación responsable es la repetición de la tripleta CTG. En el caso de la enfermedad de Huntington la mutación responsable consiste en la repetición de la tripleta CAG. Así como estas enfermedades, hay muchos otros padecimientos que son causados por tripletas repetidas.
La enfermedad de Huntington es una enfermedad genética hereditaria y rara del cerebro. Lo que causa la enfermedad es una mutación en el gen HTT (que contiene las instrucciones para producir la proteína huntingtina), localizado en el cromosoma 4. Específicamente, la mutación responsable consiste en tripletas de CAG que se repiten varias veces, más de lo normal”.
-¿Cuántas repeticiones deben existir para que se considere que una persona tiene la enfermedad de Huntington? ¿Cuánto suele ser lo normal en una persona sana?
MVC: “La población normal (sin la mutación) tiene menos de 35 repeticiones CAG. Normalmente la tripleta CAG se repite de 10 a 35 veces. Ahí no pasa nada, todo está bien.
Pero resulta que en las personas con la enfermedad de Huntington se ha visto que se repite de 36 a más de 100 veces. Las personas que tienen entre 36 a 39 repeticiones de CAG (tamaño intermedio) pueden o no llegar a tener la enfermedad de Huntington, mientras que quienes tienen 40 o más repeticiones tienen la enfermedad o van a desarrollar esta enfermedad en un futuro”.
-¿Y qué hace esta tripleta CAG para que la enfermedad se empiece a desarrollar en el paciente?
MVC: “Esa tripleta CAG da las instrucciones para generar el aminoácido glutamina. Aquí hay que repasar cómo se generan las proteínas. La información contenida en el ADN pasa a otra molécula llamada ARN y, del ARN, se generan las proteínas. Las proteínas son moléculas formadas por aminoácidos que sirven para dirigir los procesos vitales.
Entonces, ¿qué va a pasar cuando se vaya a formar la proteína huntingtina, si existe la presencia de la mutación inestable? Que esa proteína va a tener una cola muy larga, por así decirlo. O sea, un montón de glutaminas extra que van a provocar una proteína tóxica derivada del aumento en esas tripletas, mismas que se repiten más veces de lo normal.
No se sabe la función exacta de esta proteína, pero parece ser importante para las células nerviosas (neuronas) del cerebro. Así, la proteína al estar alterada va a cambiar sus interacciones y ganar una función tóxica. Al ser más larga, la proteína puede atrapar a otras proteínas y formar agregados. Esas otras proteínas se quedan ahí atrapadas sin poder cumplir su función normal”.
-¿Atrapar a otras proteínas? Básicamente, ¿no es solo que la mutación inestable genera un exceso de una proteína tóxica, sino que también captura otras proteínas que no podrán cumplir su función?
MVC: “Sí. Esos agregados se van acumulando en las neuronas. Además, la proteína mutada también se puede cortar en fragmentos tóxicos. Dichos fragmentos también se unen y forman esos conglomerados que terminan interrumpiendo muchísimas funciones normales en las células. Todos esos cambios que ocurren a nivel celular y van a provocar la sintomatología típica de la enfermedad en sus tres grupos: alteraciones motoras, de comportamiento y cognitivas. Eso sí, no afecta a todas las células del cerebro”.
-Si no afecta a todas las células del cerebro, ¿cuáles son las células neurológicas más afectadas por la enfermedad?
MVC: “Se ha visto que afecta principalmente a unas células nerviosas llamadas neuronas espinosas en los ganglios basales del cerebro. Ciertas regiones específicas de los ganglios basales son las que presentan neurodegeneración o, mejor dicho, donde mueren estas neuronas. Esta región de los ganglios basales tiene que ver con los movimientos. De ahí lo lógico es que así sea la manifestación de los pacientes que tienen problemas con los movimientos.
La enfermedad es progresiva y las células se van degenerando con el paso del tiempo. Los síntomas son cada vez más graves y es una enfermedad que dura mucho tiempo. Desde que se da el inicio de los síntomas, la esperanza de vida es de unos 15 a 20 años”.
-¿Hay algún elemento externo (ambiente, por ejemplo) que condicione la aparición genética o es exclusivamente hereditario?
MVC: “Esta enfermedad es un trastorno monogénico; o sea, causado específicamente por una mutación en un único gen. En la enfermedad de Huntington hay estudios relacionados con otro tipo de factores no genéticos como los ambientales, pero esos factores externos no son los que determinan la presencia o no de la enfermedad, aunque sí podrían influir en su progresión.
Por el momento se sabe que la presencia de la expansión de la tripleta CAG es suficiente y necesaria para el desarrollo de la enfermedad; algo muy distinto al Parkinson y al Alzheimer que son enfermedades complejas y multifactoriales, que tienen componentes tanto genéticos como ambientales.
Asimismo, más allá de las 40 tripletas, la variación en el número de repeticiones es el principal determinante de la edad a la que se inician los síntomas de la enfermedad. No obstante, se ha observado que algunos pacientes no cumplen esta pauta y sus síntomas aparecen antes o después de lo que se estimaría según el número de repeticiones de CAG que presentan. Esto ha llevado a pensar que deben existir otros factores genéticos modificadores del inicio de la enfermedad y que podrían afectar su progreso antes de la aparición de los síntomas”.
Curiosidad histórica
Usted compartió antes un dato curioso y es que esta enfermedad también se le puede encontrar bajo el nombre de corea de Huntington. También, bajo el nombre de "el mal o baile de San Vito", ¿por qué?
MVC: “El término ‘coreano’ proviene de una palabra griega que significa danza o coreografía. Esto es debido a que uno de los síntomas más frecuentes en este tipo de pacientes son movimientos involuntarios. Ellos realizan una serie de movimientos sin propósito que van fluyendo de una parte del cuerpo a otra.
Es como si estuvieran haciendo un cierto baile no planeado. Todo es completamente involuntario y de ahí el término corea. No obstante, eso se ha ido dejando de lado porque no todos los pacientes presentan ese síntoma de corea. En muchos casos infantiles la corea no es tan evidente. En cambio, sí presentan más alteraciones de carácter psiquiátrico tipo conductual o cognitivo.
Por eso se ha ido dejando de lado utilizar corea de Huntington. El otro término es mal o baile de San Vito. Anteriormente, a quienes padecían de esa enfermedad se les acusaba de estar poseídos y que tenían alguna presencia de espíritu maligno. Entonces, la gente los encomendaba a San Vito, el santo venerado como el auxiliador de las enfermedades raras”.
Aporte
-Melissa, con el aporte del Inisa-UCR en general, el país logra pasar del diagnóstico clínico al molecular de manera formal ¿Es correcto?
MVC: “Como mencioné anteriormente, antes del 2004 en el país no se hacía diagnóstico molecular de esta enfermedad. Fue hasta que se implementó en el Inisa-UCR que la enfermedad se puede diagnosticar con métodos de laboratorio. No es que ahora el diagnóstico sea molecular únicamente. El diagnóstico clínico se sigue haciendo por parte de los médicos neurólogos que tratan ese tipo de pacientes y enfermedades.
Podemos decir que el diagnóstico de la enfermedad se basa tanto en los hallazgos clínicos como en los hallazgos moleculares. Actualmente se dispone de este complemento.
El médico hace las evaluaciones clínicas, exploraciones físicas, ve la historia médica del paciente y también si existen otros casos en la familia. Él o ella pueden enviar una serie de exámenes de neuroimágenes como la resonancia magnética y la tomografía computarizada del cerebro para auxiliar en el diagnóstico. El diagnóstico se sospecha si hay señales y síntomas de la enfermedad, como las alteraciones progresivas de los movimientos, alteraciones mentales y de comportamiento.
Al tomar en cuenta todo eso, el médico en la mayoría de las ocasiones da un diagnóstico clínico de sospecha. Ahí es cuando venimos nosotros con el complemento: el diagnóstico molecular”.
-Con todo lo que usted explicó antes, se entiende que el diagnóstico molecuar es contar las tripletas CAG para saber si se llega a una cantidad que indique la presencia o no de la mutación y por lo tanto desarrollar el padecimiento.
MVC: “El diagnóstico molecular es un procedimiento que, mediante la utilización y aplicación de técnicas especializadas de biología molecular, nos permite contar ese número de repeticiones de trinucleótidos CAG. Nosotros vamos a analizar específicamente la región del gen HTT y a contar las repeticiones. El resultado del diagnóstico molecular es en términos de ese número.
Así, le decimos a la persona, por ejemplo, ‘usted tiene 30 repeticiones de la tripleta CAG en el gen HTT’. En términos sencillos, se puede decir que si una persona tiene menos de 35 repeticiones, no presenta la enfermedad. Quienes tienen más de 35 poseen o van a desarrollar la enfermedad de Huntington”.
-¿El número de tripletas que se encuentren en una persona inciden en la severidad de la enfermedad?
MVC: “Lo que se ha visto a nivel mundial es que, a mayor número de repeticiones CAG presentes en la persona, la enfermedad se manifiesta a edades más tempranas y con síntomas más severos. Este fue el caso de una niña en la que encontramos que tenía 125 repeticiones.
Por su parte, entre menor número de repeticiones de las tripletas, la enfermedad se manifiesta a edades más tardías y con síntomas más leves. Por lo general, esta enfermedad posee una manifestación entre los 30 a los 50 años, edades en las que muchas personas ya han formado sus familias y han tenido hijos”.
-En el caso de Costa Rica, ¿cuál es el número promedio de tripletas que ustedes han encontrado?
MVC: “La mutación en promedio anda en 45 repeticiones. Hemos identificado tres casos que tienen 55 de esas tripletas y que coinciden en presentar los síntomas de la enfermedad a edades tempranas (20-25 años).
También hay casos de inicio tardío que empiezan a manifestar síntomas después de los 55 o 60 años. Son personas que están en el umbral más bajo (entre 35 a 40 repeticiones) con síntomas leves como ‘tics’ que suelen confundirse con Parkinson o como consecuencia del propio envejecimiento”.
-Desde que inicia la aplicación de esta prueba molecular, ¿a cuántos pacientes han atendido a lo largo del tiempo en el Inisa-UCR?
MVC: “Del 2004 a la fecha, en el Inisa-UCR hemos diagnosticado molecularmente a 162 personas. De esos 162, 71 personas tenían un diagnóstico clínico o sospechoso de Huntington referenciado por un neurólogo. De esos 71 individuos, la mutación se identificó únicamente en 42 pacientes. Los demás tenían síntomas similares pero no esa enfermedad.
Las 91 personas restantes correspondían a familiares en riesgo (tenían a un pariente con la enfermedad de Huntington). Las personas con antecedentes en su familia poseen un riesgo del 50 % de heredar o haber heredado la mutación, es como tirar una moneda al aire: es escudo o es corona. De esos 91, solamente 28 resultaron ser positivas (portadoras de la mutación) y todavía no presentaban síntomas al momento del estudio.
Para resumir, a la fecha hemos identificado 70 personas con la mutación, 43 son mujeres y 27 son hombres. De esas 70 personas alrededor de 20 ya fallecieron. Por lo tanto, en el país habría unas 50 personas vivas diagnosticadas a nivel molecular por el Inisa-UCR”.
, lo hacen para orientar su futuro familiar de tener o no hijos.")
Muchos de los que se hacen la prueba presintomática (personas sin síntomas pero en riesgo), lo hacen para orientar su futuro familiar de tener o no hijos.
-Melissa, ¿hay alguna provincia o zona geográfica del país en donde se presente más este padecimiento?
MVC: “No. La distribución está por todo el país. Hay muchos que están en el valle central y la mayoría se concentran en tres familias que tienen el mayor número de afectados. Nosotros no hemos hecho un estudio genealógico en profundidad, pero es posible que varias de estas familias tengan un ancestro en común.
Hemos encontrado que el número de repeticiones de la tripleta coinciden con lo visto a nivel mundial. La mayoría de las mutaciones andan entre los 40 y las 55 repeticiones, excepto un caso que encontramos con 125 repeticiones de la tripleta en una niña de seis años. Ella fue el primer reporte de un caso infantil con diagnóstico molecular en el país".
-¿Es el diagnóstico temprano una opción para retardar la aparición de esta enfermedad?
MVC: “No. No hay un tratamiento curativo disponible para la enfermedad. En el campo clínico lo que se hace es abordar los síntomas mediante medicamentos específicos para tratar los movimientos, medicamentos para tratar los síntomas de depresión o psiquiátricos. En resumen, en eso consiste la intervención farmacéutica actual.
No hay un medicamento aprobado o alguna terapia que modifique la progresión de la enfermedad. Existen terapias o tratamientos para síntomas que brindan un alivio a las personas afectadas. Aquí lo que se busca es que tengan la mejorar calidad de vida posible”.
-¿Cuáles son los tratamientos actuales para esta enfermedad?
MVC: “Las personas pueden optar por tratamientos como fisioterapia para la parte motora, terapias verbales y terapias también relacionadas con la masticación y deglución porque los movimientos de los pacientes son irregulares. Asimismo, hay terapias ocupacionales para adecuar sus casas y sus lugares de trabajo.
Por otro lado, en la actualidad hay muchas investigaciones y estudios clínicos en proceso dirigidos a tratar de disminuir los niveles de la proteína tóxica que origina la enfermedad; es decir, intentar silenciar esa proteína para detener la presentación del padecimiento.
No obstante, aún no se ha logrado. Por lo tanto, como le indiqué antes, en estos momentos el tratamiento se basa en tratar los síntomas con fármacos y con campos multidisciplinarios que involucran nutricionistas, terapeutas físicos, terapistas ocupacionales y hasta trabajadores sociales por toda la problemática que esta enfermedad trae. No es solo una persona con la enfermedad, sino que involucra a todo el núcleo familiar porque requiere muchos cuidados en sus etapas más avanzadas”.
Puertas abiertas
-¿Qué necesitan ustedes del paciente para hacer el diagnóstico molecular y cómo es el proceso?
MVC: “Necesitamos tomar una muestra de sangre del brazo, como si fuera a realizarse un examen a cualquier laboratorio clínico. Nosotros hacemos la toma de la muestra y la procesamos. Para procesarla primero rompemos las células y luego, mediante una serie de procedimientos en el laboratorio, obtenemos solamente el material genético (el ADN).
Una vez con el ADN se hace una amplificación y usamos la técnica de reacción en cadena de la polimerasa (PCR) para generar múltiples copias de ese material genético, específicamente, de la región donde se ubica el gen HTT que a nosotros nos interesa: el responsable de la enfermedad de Huntington que está en el cromosoma cuatro.
Una vez que yo tengo eso, confirmo que la PCR se llevó a cabo correctamente, pero no puedo contar el número de repeticiones. Por eso, mandamos las muestras ya purificadas listas al Centro de Investigación en Biología Celular y Molecular (CIBCM-UCR) con la doctora Ivannia Atmetlla.
Ahí ella corre las muestras en un equipo llamado secuenciador y lo que obtenemos es otro tipo de resultados que se llaman electroferogramas, que son unas oscilaciones (unos picos) en diferentes colores que me van indicando la secuencia del segmento que amplificamos. Mediante los electroferogramas se analiza la secuencia y vamos contando cuántas tripletas tiene esa persona”.
-¿Cómo una persona interesada en conocer si tiene o no la enfermedad puede llegar a ustedes?
MVC: “Las personas que solicitan la prueba son por lo general pacientes que tienen una referencia de un médico neurólogo y, en otros casos, son familiares en riesgo; personas que ya tienen antecedentes de Huntington en su familia.
En cualquiera de los dos casos, solo llaman al Inisa-UCR y sacan una cita. Una vez en el INISA, hacemos una entrevista para obtener información personal y los antecedentes familiares, se les lee un consentimiento informado y si están de acuerdo con realizarse el examen se procede a la firma del consentimiento. Durante todo el proceso intentamos emplear siempre un lenguaje sencillo para que el paciente entienda y le damos mucho material para que sepa en qué consiste la prueba.
Con la explicación, el paciente puede elegir si se hace o no la prueba. Algo muy importante es que con los pacientes siempre intentamos ser muy claros de que no hay una cura para su enfermedad. Siempre intentamos ser muy sinceros porque muchos vienen con esa esperanza.
Algunos si vienen más al tanto de qué es la enfermedad, pero hay otros casos un poco más difíciles porque nunca han oído hablar del padecimiento. Ni siquiera sabían o simplemente llegaron donde el médico y por la sintomatología que presentaban se les dio un diagnóstico clínico de Huntington. A veces resulta ser que son el primer caso del núcleo familiar porque no han tenido mucha relación con otros miembros familiares. Otros desconocen que es algo hereditario y que sus hijos e hijas también pueden tenerlo”.
-¿Hay algún cambio en la recepción de pacientes por el contexto de la pandemia?
MVC: “Desde marzo del año pasado (2020) no hemos estado recibiendo pacientes por la situación de la pandemia. No obstante, hemos estado montando una lista y ya tenemos a 20 pacientes que contactaremos tan pronto como sea posible. Si hay otras personas interesadas, se pueden ir sumando a esta lista perfectamente”.
-¿La prueba tiene algún costo? ¿Cuánto tardan ustedes en dar los resultados?
MVC: “El costo de la prueba es de 100 dólares al tipo de cambio vigente, y solo cubre los costos de los reactivos que se necesitan para hacer las pruebas, no el recurso humano porque este lo da la UCR. En cuanto a los resultados, estos se dan en aproximadamente dos meses. Nosotros tratamos de procesar varias muestras juntas por cuestiones de protocolo en el laboratorio, ahorro de tiempo y de consumibles.
Por eso, en ocasiones no procesamos una muestra sino que nos esperamos a tener varias. En caso de que ya se esté tardando mucho en tener un grupo de muestras procedemos a procesar las que tenemos para que los pacientes no esperen tanto tiempo, ante una situación que genera gran angustia, incertidumbre y preocupación”.
Ventajas del diagnóstico molecular
1. Identificar a personas con enfermedad y familiares en riesgo.
2. Clasificar de forma correcta a los pacientes y dar un mejor manejo clínico.
3. Disminuir la recurrencia de personas afectadas en el país.
4. Aportar en futuras terapias al tener identificados este tipo de pacientes para estudios futuros o tratamientos.

Periodista, Oficina de Divulgación e Información
Área de cobertura: ciencias de la salud
jenniffer.jiarvtmenezcordoba @ucrchls.ac.cr
Comentarios:
Artículos Similares:
-
 Dr. Molina: “Costa Rica está entre los países con los niveles más graves de bacterias resistentes”
Dr. Molina: “Costa Rica está entre los países con los niveles más graves de bacterias resistentes” -
 Voz experta: El Centro de Investigación en Cirugía y Cáncer (Cicica-UCR) innova la investigación …
Voz experta: El Centro de Investigación en Cirugía y Cáncer (Cicica-UCR) innova la investigación … -
 ¿Por qué después de varios años hablamos de nuevo sobre la tosferina? El Dr. Rojas lo explica
¿Por qué después de varios años hablamos de nuevo sobre la tosferina? El Dr. Rojas lo explica -
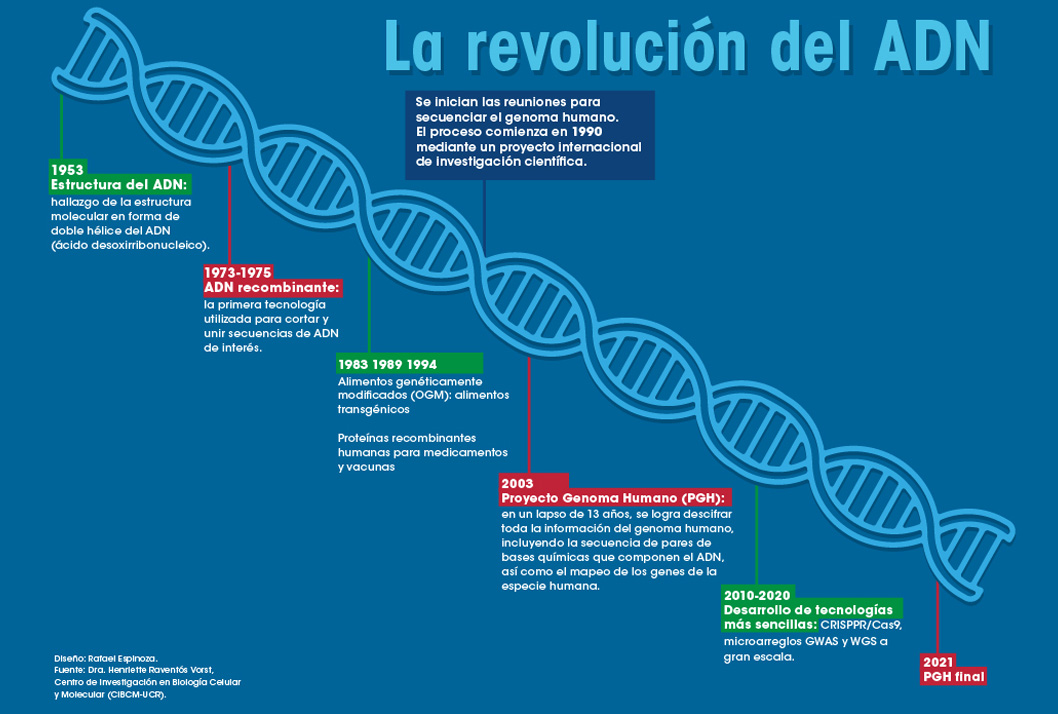 A 70 años del descubrimiento de la estructura del ADN
A 70 años del descubrimiento de la estructura del ADN
